- Visibility 49 Views
- Downloads 29 Downloads
- DOI 10.18231/j.ijpns.2024.014
-
CrossMark

The droopy eyed toddler – A rare case of juvenile myasthenia gravis
Introduction
Myasthenia gravis is one of the differential diagnosis to be considered in a child presenting with muscle weakness. Basic pathology here is abnormal Acetylcholine release from Neuromuscular Junction so that prior binding of Acetylcholine to post synaptic terminal does not occur resulting in abnormal muscle function. Immune mediated variety is associated with auto-antibodies attached to post synaptic terminal causing degradation of Acetylcholine receptor. This can result in weakness and early fatigability.[1]
Different pathophysiological mechanisms are associated with the 3 distinct forms of Myasthenia Gravis affecting children. The Congenital Myasthenic Syndromes (CMS) are a group of genetic disorders that lead to muscle weakness through structural or functional abnormalities of the proteins that are involved in neuromuscular transmission. Trans placental transfer of maternal antibodies from a myasthenic mother causes infants to have Transient Neonatal Myasthenia (TNM). Autoimmune mediated dysfunction of Acetylcholine receptors( AChR) results in Juvenile Myasthenia Gravis (JMG). All 3 forms of myasthenia causes muscle fatigue and weakness of varying degrees.
Myasthenia gravis is a rare disease in paediatric population with a detection rate of only 1 in 1 million cases; rates are found to be even lower in pre pubertal children. [2]
Case Presentation
18 months old boy, with normal growth and development and no significant past illness, presented with complaints of difficulty in opening eyes for the past 1 month. There was a history of viral prodrome 1 week before OPD visit. He had no associated difficulty in swallowing, speaking or limb weakness initially. No altered sensorium, vomiting, seizures or fever at time of presentation.
Ophthalmology consultation was taken to rule out inflammatory conditions of eyes. He was prescribed steroid eye drops for 2 weeks considering possibility of viral conjunctivitis associated post inflammatory edema. On review after two weeks, not only was there any improvement but there also was diurnal variation in eyelid drooping. Additionally, there was lack of facial expressions, cough while eating, swallowing difficulty especially for liquids and frequent falls in the past 2 week’s duration. There was no family history of any neurological illness. On examination child was active and alert with bilateral ptosis. His vitals were stable. Neurological examination showed Grade 5 Power of bilateral upper and lower limbs, tone and reflexes were normal. Meningeal and cerebellar signs were absent. Rest of CNS and other system examination was within normal limits. Suspecting myasthenia, ice pack test was done, which turned out to be positive.
Child was referred to Neurologist to to confirm the diagnosis of Myasthenia gravis. Neostigmine challenge test was done, which showed significant improvement in ptosis, thereby confirming myasthenia. Repetitive nerve stimulation test was not done as diagnosis was confirmed with neostigmine test and considering the painful nature of the test.
Blood investigations showed normal blood counts, CRP, TSH, LFT and Serum electrolytes. No mediastinal mass was detected on Chest X-ray and Ultrasound abdomen was normal. ECG and ECHO done to rule out any cardiac involvement was found negative. Anti Ach Receptor antibody, Anti MUSK antibody, ANA Profile was Negative. Whole Exome Sequencing to look for mutations suggestive of congenital myasthenic syndromes came negative. Child was diagnosed to have double seronegative, possibly immune mediated myasthenia gravis. He was managed with Tab. Pyridostigmine in 4 divided doses and oral steroids, with which the child showed drastic improvement. Child did not develop any further complications like respiratory paralysis. Presently, his symptoms are well controlled and kept under regular follow up.
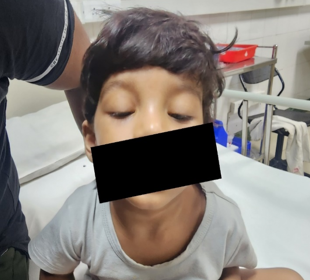
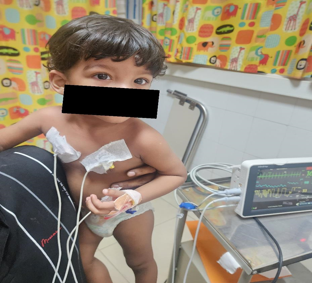
Discussion
Myasthenia gravis is is potentially life threatening neuromuscular junction disorder in which there is fatigue of the musculature.[3] Myasthenia gravis presenting in pre pubertal age group is relatively rare. 75% Children present after the age of 10 years. Pre pubertal type is associated with a male bias, ocular symptoms only and seronegativity for AchR antibody. 40-50% can have weakness of other bulbar and limb muscles.
Immune mediated juvenile myasthenia gravis usually presents in paediatric age group and is not a transient condition nor is it due to structural disorder that causes congenital myasthenia syndrome.[4] Here it is important to rule out other associated autoimmune conditions like thyroiditis/polymyositis. Congenital myasthenia syndrome comprises of a constellation of signs and symptoms which can present from birth to early childhood. Usual early symptoms are ptosis and weakness. Due to early presentation, other congenital myopathies and muscle dystrophies should be ruled out. [5] Here defect maybe in structure of neuromuscular junction, acetylcholine receptor or in the end plate. Detailed genetic analysis is pertinent to reveal the pathology in this patient. The main pillar of diagnostic workup in immune mediated myasthenia gravis is AchR antibody testing. Binding, blocking and modulating antibodies to AchR are tested. [6] Patients who are negative to AchR antibody or anti MUSK antibody are labelled as double seronegative Myasthenia gravis.
Clinical diagnosis can be accomplished by administration of Edrophonium or Neostigmine which are fast acting cholinesterase inhibitors. Edrophonium prevents break down of acetylcholine and increases concentration of acetylcholine at neuromuscular junction causing improvement in patient’s muscle function. Careful monitoring during the test is important as child can develop bradycardia, nausea, hyper salivation or cholinergic crisis. Repetitive nerve stimulation using low frequency stimuli can also be done. A 10% decrementation between 1st and 5th wave is considered pathognomic. [3]
Cogan lid twitch test is a simple out patient based procedure whereby a period of rest will bring back muscle function to normal, thereby ameliorating ptosis. 2 mm improvement in ptosis following ice pack placement over eyelid is another diagnostic point in favour of myasthenia gravis. Sensitivity of this is 80%. [7]
First-line treatment of this condition is administration of long acting acetylcholine esterase Pyridostigmine 7mg/kg/day in 4-6 divided doses.
Second line agents often used as add on therapy for immune modulation includes Azathioprine (a purine analogue that suppresses B and T cell proliferation). Multiple other immunomodulatory agents like Rituximab, Eculizumab, Mycophenolate mofetil and Tacrolimus have been used in MG to control symptomatology.
Another therapeutic modality in antibody positive patients is surgical removal of thymic germinal centre which will disrupt auto antibody production leading to improvement of symptoms or induction of remission. [8]
Plasmapheresis and intravenous immune globulin (IVIG) are useful in management of myasthenia gravis or in priming patient prior to thymectomy. [9]
Complicated myasthenia gravis management requires a multidisciplinary team comprising a paediatrician with help from paediatric neurologist, physiotherapist, occupational therapist, psychologist, speech therapist and dietician.
Conclusion
Myasthenia gravis is a disorder that can lead on to life threatening respiratory arrest and ventilator dependence which causes significant morbidity and mortality in the patient. Hence early detection and proper treatment of the condition is very essential. Though uncommon in prepubertal age group, when a child presents with bilateral ptosis a clinical suspicion of myasthenia has to be considered as it may be an early presentation of the disease. Detailed history taking and clinical examination followed by relevant investigations is the mainstay of diagnosis. Early initiation of treatment is vital in preventing complications of the disease. Early diagnosis and effective treatment has significantly improved the long term outcomes with reduction in morbidity and mortality in these patients.
Source of Funding
None.
Conflict of Interest
None.
References
- K O’connell, S Ramdas, J Palace. Management of Juvenile Myasthenia Gravis. Front Neurol 2020. [Google Scholar] [Crossref]
- JH Peragallo. Pediatric Myasthenia Gravis. Semin Pediatr Neurol 2017. [Google Scholar]
- PI Andrews. Autoimmune myasthenia gravis in childhood. Semin Neurol 2004. [Google Scholar]
- A Evoli. Acquired myasthenia gravis in childhood. Curr Opin Neurol 2010. [Google Scholar]
- M Kinali, D Beeson, MC Pitt, H Jungbluth, AK Simonds, A Aloysius. Congenital myasthenic syndromes in childhood: diagnostic and management challenges. J Neuroimmunol 2008. [Google Scholar] [Crossref]
- WKM Liew, PB Kang. Update on juvenile myasthenia gravis. Curr Opin Pediatr 2013. [Google Scholar]
- K C Golnik, R Pena, A G Lee, E R Eggenberger. An ice test for the diagnosis of myasthenia gravis. Ophthalmology 1999. [Google Scholar]
- A Hayashi, H Shiono, M Ohta, K Ohta, M Okumura, Y Sawa. Heterogeneity of immunopathological features of AChR/MuSK autoantibody-negative myasthenia gravis. J Neuroimmunol 2007. [Google Scholar]
- P Gajdos, S Chevret, K V Toyka. Plasma exchange for generalised myasthenia gravis. Cochrane Database Syst Rev 2002. [Google Scholar]